Natural History



SMA is a rare, progressive neuromuscular disease1,2


Please note, throughout this website, when discussing “SMA”, we are referring to 5q SMA, the most common form of the disease.
SMA (5q SMA) is a rare, progressive neuromuscular disease caused by the degeneration of motor neurons in the spinal cord, which results in skeletal muscle atrophy and weakness.1,2
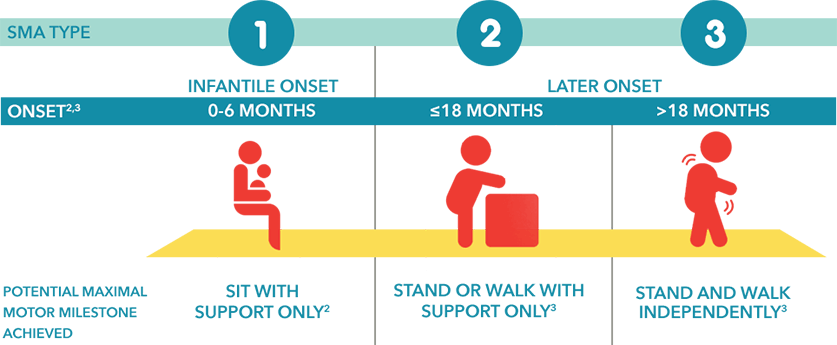
Each individual with SMA is unique. SMA can present with a wide spectrum of symptoms and is often classified by clinicians based on the age of onset, severity of symptoms, and disease progression.2
In individuals with untreated SMA, the prognosis is often related to maximum motor function achieved.6