Genetic Cause



The role of SMN1 gene in SMA


Please note, throughout this website, when discussing “SMA”, we are referring to 5q SMA, the most common form of the disease.
SMA (5q SMA) is caused by a mutation or deletion of the SMN1 gene on chromosome 5q.1
- In healthy individuals, SMN1 is the primary producer of functional, full-length SMN protein;2
- In individuals with SMA, the missing SMN1 gene results in levels of functional, full-length SMN protein that are insufficient to sustain the survival of motor neurons;2-5
- At the time of the discovery of the SMN1 gene, a duplicate gene, now known as SMN2, was also found.6
Functional, full-length SMN protein is necessary to maintain the health and function of motor neurons in the spinal cord.5

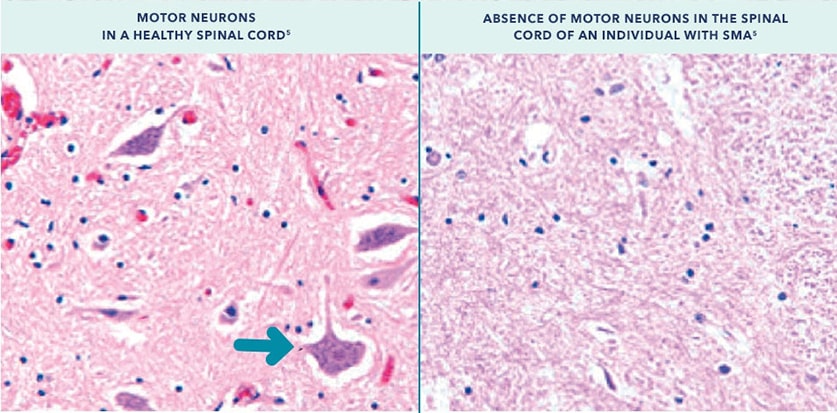
Arrow indicates a healthy motor neuron.
With permission from Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120-2133.
How does untreated SMA manifest?
LEARN MORE
Learn more about the natural history of untreated SMA.